Department of Biomedical and Chemical Engineering, Syracuse University, Syracuse, New York 13244, United States
More by Saeid Biria, Shreyas Pathreeker, Hansheng Li, and Ian D. Hosein*
Cite this: ACS Appl. Energy Mater. 2019, 2, 11, 7738–7743
Publication Date:October 30, 2019
Abstract
We report the plating and stripping of calcium metal at room temperature in a mixture of ethylene carbonate (EC) and propylene carbonate (PC) solvents using calcium tetrafluoroborate (Ca(BF4)2) as the salt. Calcium is reversibly deposited and removed over several cycles, with associated plating–stripping efficiencies of >95%. Thin layers (∼20 μm) of crystalline calcium are present as the deposits, with a stable solid electrolyte interface (SEI) forming. This work opens opportunities to realize redox active Ca metal electrodes using commercially available alkyl carbonate solvents.
Introduction
Stable and efficient redox reactions are a critical aspect to electrochemical systems, in order to support, as examples, efficient energy conversion and storage, materials synthesis, and gas/liquid phase analyte detection. Particularly for energy storage, the push to introduce pure metals as anodes in electrochemical systems relies on effective redox-based deposition and dissolution of metal for practical delivery and storage of electrical energy. This has led to a significant body of work resulting in the successful plating (reduction) and stripping (oxidation) of different metals, particularly those of interest being lithium, sodium, and magnesium. (1−3) Furthermore, in the context of practical energy systems, plating and stripping under ambient conditions (specifically at room temperature) are critical and have thus far been achieved with the above metals, among others.
Practical, efficient plating and stripping of calcium metal at room temperature is a critical aspect in the development of calcium metal electrodes, motivated especially by calcium batteries as a promising next-generation electrochemical energy storage system. (4) Among its many advantages, calcium is the fifth most abundant mineral in the Earth’s crust. A calcium metal anode offers significantly higher volumetric and gravimetric electrochemical capacities (2072 mAh mL–1 and 1337 mAh g–1, respectively) than current commercial graphitic anodes in Li-ion batteries (300–430 mAh mL–1 and 372 mAh g–1). (3) Calcium metal anodes have a 2+ oxidation state which provides greater energy density over monovalent systems (i.e., Li+ and Na+) and a standard reduction potential 0.17 V greater than that of Lithium. Plating of calcium metal does not suffer from dendritic growth, which has plagued lithium metal electrodes and inhibited their practical use. (4) As compared to divalent systems, calcium-ion batteries would have a higher cell voltage than Mg2+ because of the 0.5 V lower standard reduction potential of Ca/Ca2+ vs Mg/Mg2+. Ca2+ also has the potential for faster reaction kinetics as compared to Mg2+ owing to its lower polarizing properties and charge density.
However, practical redox reactions associated with plating and stripping of calcium metal at room temperature in a suitable electrolyte have proven difficult to achieve, consequently requiring elevated temperatures to obtain sufficient redox kinetics or otherwise leading to side reactions and significant electrolyte decomposition. Initial studies by Aurbach et al. on calcium plating/stripping in several organic electrolytes deemed it impossible, owing to the lack of Ca2+ diffusion across formed passivation layers. (5) Hayashi et al. also found that plating/stripping of calcium was not possible in several organic electrolytes. (6) More recently, Ponrouch and Palacin reported the reversible plating and stripping of calcium in carbonate solvents, but only above 75 °C (and with improvement at higher temperatures up to 100 °C) and accompanied by significant quantities of a secondary CaF2 phase due to electrolyte breakdown. (7) No redox reactions at room temperature could be observed. Wang et al. reported the deposition of calcium in a tetrahydrofuran (THF) solvent; however, significant electrolyte decomposition produced CaH2, which formed a passivating layer inhibiting Ca2+ transport, thereby terminating plating/stripping kinetics. (8) This confirmed earlier work on THF solvents, which also observed difficulty with eliciting redox reactions. (5) Hence, reversible room temperature plating and stripping in suitable, stable electrolytes remains an open issue in the electrochemistry of calcium. (4,9) Thus far, conventional polar aprotic solvents, such as alkyl carbonates, appear the most promising candidates as indicated in preliminary experimental and theoretical work, (3,7,10,11) as well as owing to and motivated by their success in Li batteries. (12) However, despite this potential, practical room temperature calcium plating and stripping reactions in alkyl carbonate electrolytes have not been reported.
Herein we report the plating and stripping of calcium metal at room temperature in an electrolyte consisting of a mixture of ethylene carbonate and propylene carbonate (EC-PC) containing 1 M calcium tetrafluoroborate (Ca(BF4)2) as the salt. An EC-PC solution is a common carbonate-based electrolyte mixture, used in lithium-ion batteries, that is also promising for use with calcium. (9) EC is known to form stable, ion-permeable passivation layers and, when mixed with PC (which is nonreactive with Ca (5)), forms a stable liquid solvent, and their combination possesses a wide liquidus range and electrochemical stability window. Ca(BF4)2 salt is attractive owing to its capability to readily solvate and desolvate from alkyl carbonate molecules (9) and its contribution to the formation of stable ion-permeable passivating layers, (10) as well as for the stability of the BF4– anion. We show the capability to deposit and remove thin (∼20 μm) calcium metal films on a copper (Cu) substrate over 20 cycles. We selected Cu owing to its stability at low potentials, which makes it suitable as a potential current collector for a calcium metal electrode. This is a critical step toward realizing a practical calcium metal electrode redox reaction and opens opportunities to explore calcium redox processes in other alkyl carbonate electrolytes as well as advance studies such as for the development of calcium batteries.
Results and Discussion
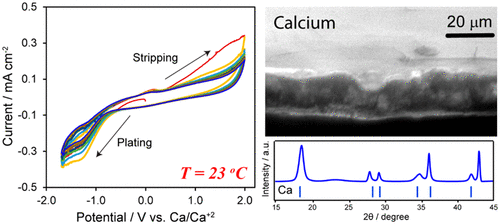
Figure 1 shows cyclic voltammetry (CV), galvanostatic (GS) cycling, and electrochemical impedance spectroscopy (EIS) data for calcium plating and stripping, particularly for the first, fifth, and 10th cycles (three cycles shown for clarity). Measurements were performed within the oxidative stability window of the electrolyte (∼3 V). (10) A redox process is observable at room temperature. Namely, voltammograms are characteristic of reversible metal plating and stripping. The onset potential difference was found to be 0.95 V for the fifth and 10th cycles, and the smallest difference between oxidation and reduction was 0.65 V in the first cycle. Plating and stripping potentials were −1.2 and 0.25 V vs Ca/Ca2+, respectively, and were consistent over all cycles. The reversible redox reactions were observed up to 20 cycles (to demonstrate reliable capability for room temperature plating and stripping). CV data for all other cycles are provided in the Supporting Information. GS cycling investigated plating and stripping up to areal capacities of 2.2 mAh cm–2. Plating–stripping cycle efficiencies were >95%, indicating the stability and reversibility as a potential anode operating at room temperature. Extracted nucleation overpotentials from the GS plating curves were in the range of −10 to −340 mV (see the summary in the Supporting Information). The increase in the stripping potential with cycle number is most likely associated with the formation of the SEI layer, owing to bulk and grain boundary resistances, which begin to stabilize the stripping potential by the 10th cycle. Nyquist plots of the impedance spectra before plating/stripping and after the first and fifth cycles (data for all other cycles provided in Supporting Information) show that the initial Cu surface begins with ohmic resistance and a semicircle at lower frequencies. The semicircle corresponds to processes at the interface associated with the SEI layer and charge-transfer resistances of the metal electrode. (13) The resistance at the electrolyte interface (RSEI) was on average 702 and 360 Ω, after plating and stripping respectively. RSEI before cycling was found to be ∼440 Ω. A summary of RSEI values after each cycle can be found in the Supporting Information. Values on this order of magnitude indicate suitable cation permeability associated with metal plating and stripping and are similar to those reported for SEI layers in lithium plating and stripping. (14) The resistance at the electrolyte interface after plating is much higher than that after stripping for all cycles, which is in accordance with the directly proportional relationship between RSEI and SEI thickness, (15) as the stripped layer is thinner than the plated layer, which is confirmed by SEM images (vide infra). RSEI decreases initially with cycle number, possibly indicating an increase in ionic conductivity within the SEI layer, as has been reported previously for other systems. (16−18) Then RSEI gradually stabilizes during subsequent cycles and remains at a value higher than that before cycling, which points toward formation and retention of a stable SEI layer. (19)

Figure 2 shows the characterization of the calcium deposits as well as the Cu substrate, after polarization at room temperature for the first and 10th GS cycles (constant current of 0.25 mA for 4 h). XRD spectra indicate diffraction peaks that correspond to reference calcium metal (structure, cubic close-packed (CCP); space group, Fm3̅m). Spectra between 15 and 45° are shown to clearly reveal the Ca XRD peaks, owing to the strong intensity peaks from the Cu substrate (full spectra provided in the Supporting Information). Upon subsequent stripping, the Ca peaks disappear, indicating the removal of calcium metal. The broad signal between 20 and 35° is associated with surface passivation by an SEI layer on the Cu substrate that forms upon the first plating step (i.e., reduction), during which calcium is deposited along with traces of side products. The persistence of this broad peak in the XRD in the stripping (i.e., oxidation) step is due to the breakdown of the plated layer due to diffusion of calcium, thereafter leaving remnant side products on the Cu surface. This is a similar phenomenon as observed for lithium metal electrodes. (20) No other crystalline phases were found, indicating minimal side reactions occurred and the primary crystalline deposit was calcium metal. Particularly, the observed XRD peaks shown in Figure 2a could not be assigned to reference CaF2. Hence, it appears that plating at room temperature avoids secondary crystalline phases, such as CaF2 observed when plating at elevated temperatures. (10) Furthermore, the electrochemical stability of EC-PC at room temperature appears to avoid the formation of secondary crystalline phases that might be produced by reactions between calcium and the electrolyte, as observed for plating/stripping in non-carbonate-based solvents, such as THF which produces CaH2. (8) EDX spectra show the appearance and disappearance of the calcium peaks (Kα = 3.69 and 4.038 keV) with the first plating and stripping, respectively, also confirming the reversible deposition and removal of calcium from the Cu substrate. The existence of a calcium peak in the spectra after the 10th stripping indicates the presence of remnant calcium, which XRD confirms to be crystalline. The EDX spectra also show the presence of carbon, oxygen, and fluorine peaks, all of which remain in the sample across plating and stripping steps. These elements constitute the solid electrolyte interface (SEI) layer formed by small amounts of electrolyte breakdown. To further compositionally understand the deposits, FTIR analysis of the Cu substrate after plating and stripping show spectra that are relatively unmodified between these two steps. The FTIR spectra reveal small amounts of EC, PC, and their constituents, all of which are associated with electrolyte and electrolyte constituents that form the SEI layer. Specifically, low intensity FTIR peaks associated with the BF4– anion may indicate trapped electrolyte in the SEI layer, which would explain the fluorine peak observed in the EDS spectra and the lack of significant CaF2 peaks in the XRD. This would also indicate that, at room temperature, the formation of CaF2 is not as prominent, at least to the extent observed at higher temperature. A summary of the wavenumbers assigned to each peak of interest can be found in the Supporting Information. The FTIR spectra were also unmodified between the 10th plating and stripping (see the Supporting Information). These results indicate that the SEI layer lacks secondary crystalline phases (i.e., from XRD analysis) and is stable and permeable to Ca2+ (i.e., from EIS analysis) owing to the capability to reversibly plate and strip calcium metal.

Figure 3 shows the morphology of the calcium deposits on the Cu substrate as well as the surface of the substrate after stripping. A thin, dense film of crystalline calcium metal deposits is present with the first plating step. The ∼20 μm thickness of the deposits is in close accordance to what may be calculated on the basis of the total charge density applied (2.2 mAh cm–2) to a 0.45 cm2 substrate, namely, 7.92 C of charge yields ∼1650 μg of deposited calcium metal, which would produce an ∼23.6 μm thick film. Upon stripping, the calcium is predominately removed (Figure 3b), and there is an apparent surface morphology (Figure 3d, inset) associated with the passivation (SEI) layer over the Cu surface. Closer images show that this layer is a continuous film (see the Supporting Information), yet it still retains the outline of the boundaries of the calcium grains that were deposited under it. This may indicate that while calcium deposition occurs through the nucleation of islands, the outlining regions of the SEI layer possibly allow for conformal coating of the grains (its surface and side grain boundaries). Yet, it is possible that Ca2+ may find cracks or other defects through which it can diffuse to reach the growing Ca deposit. Careful studies on SEI formation, morphology, and transport are the subject of current study. The calcium islands that form have the same size range, predominately from 20 to 100 μm, for both the first plating and 10th plating (Figure 3c). These observations reveal the capability to plate and strip over several cycles to obtain the repeated deposition of thin, dense crystalline calcium metal films.

The results confirm the capability to reversibly plate and strip calcium metal in an alkyl carbonate based electrolyte. One explanation for the achieved deposition and removal of calcium at room temperature could be the use of a Cu substrate, which is a suitable collector for metal electrodes owing to its stability at low potentials, as opposed to stainless steel or aluminum which are generally more suited for positive electrodes. The lack of any detectible CaF2 both in the deposits as well as in the SEI layer could also favor the capability to more easily plate and strip Ca. Herein, BF4– appears electrochemically stable at room temperature, an observation corroborated by others previously. (5) Hence, it is possible that CaF2 formation is not as predominant at room temperature, at least to the extent detectable, and is more likely to decompose at higher temperatures. Hence, the passivating nature of CaF2 may be mitigated at room temperature. While EC-PC solutions have been shown herein to be favorable, other alkyl carbonate electrolytes have been theoretically predicted to also effectively solvate and desolvate calcium, (9) and plating and stripping in other such electrolytes is currently underway and will be reported on in the future. Nevertheless, this combination of a calcium metal electrode, suitable electrolyte, and plating–stripping conditions presented herein opens opportunities to explore calcium metal electrodes in applications, as well as to consider them, for example, with new high voltage cathode materials (21−24) toward study of electrochemical energy storage.
Conclusion
We have shown the reversible plating and stripping of calcium metal on a Cu substrate at room temperature in an alkyl carbonate based mixture of EC and PC with Ca(BF4)2 salt, which provides high plating/stripping efficiency over several cycles, and stable SEI formation. This work overcomes previous challenges with plating and stripping calcium metal, which have resulted in passivating electrolyte decomposition or the need for elevated temperatures. Advantageously, practical plating (reduction) and stripping (oxidation) open opportunities to explore the use of redox-capable calcium thin films for applications in which metal electrodes play critical roles.
Experimental Section
Materials
Calcium tetrafluoroborate hydrate (Ca(BF4)2)·xH2O was purchased from Alfa Aesar. Ethylene carbonate (anhydrous, 99.0%) and propylene carbonate (anhydrous, 99.7%) were purchased from Sigma-Aldrich. The calcium salt was vacuum-dried at 120 °C overnight before use. Ethylene carbonate and propylene carbonate were used as received. Calcium metal was purchased from Goodfellow, and copper foil (∼25 μm thickness) was purchased from MTI Corp.
Electrolyte Preparation
Ca(BF4)2 salt was dissolved in a 50/50 (v/v) mixture of EC and PC as solvent to prepare a 1 M solution which was stirred for 24 h.
Electrochemistry
Cyclic voltammetry (CV) experiments were carried out in a three-electrode beaker type cell (Gamry Instruments), wherein platinum wire (Gamry) was used as the counter electrode and Ca/Ca2+ was used as the reference electrode. The scan rate was 25 mV s–1. Galvanostatic (GS) plating and stripping of calcium on the working electrode was carried out in a three-electrode type cell where pure calcium metal was used as both the counter and the reference electrodes. Cycling was performed at a constant current of 250 μA. Copper (Cu) foil of 0.45 cm2 area was used as the working electrode in the CV, GS, and EIS experiments. EIS measurements were performed over a frequency range of 0.1 Hz to 1 MHz at a fixed amplitude of 30 mV. The Cu foil was cleaned by placing over it a few drops of N-methyl-2-pyrrolidone (NMP) and then wiping it dry. EIS was performed before cycling, after plating and after stripping for eight cycles. Impedance of the interfacial layer, termed as RSEI, was extracted from the intercept of the high to middle frequency semicircle in the Nyquist plots on the x-axis. (14) CV, GS, and EIS measurements were carried out with a Solartron Energy Lab XM Instrument. All measurements were performed in an argon filled glovebox maintained at <1 ppm O2 and H2O content. All plated and stripped samples were stored in the glovebox until further characterization was performed.
Electrode Characterization
Electron microscopy was carried out with a scanning electron microscope (JEOL 5600) equipped with an energy dispersive X-ray (EDX) detector used under an accelerating voltage of 10 keV. X-ray diffraction spectra were collected using a Rigaku diffractometer. Cu Kα radiation was used to collect data in a diffraction 2θ range from 10° to 80° at room temperature. Reference XRD for Ca and Cu were acquired from the calcium metal and copper foil, respectively. FTIR measurements were performed with an ATIR-FTIR spectrometer (Bruker, Alpha), and spectra were collected in transmittance mode.
【Article link】
https://doi.org/10.1021/acsaem.9b01670