Polymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, Lund SE-221 00, Sweden
More by Zhecheng Shao
, Hannes Nederstedt, and Patric Jannasch*
Cite this: ACS Appl. Energy Mater. 2021, 4, 3, 2570–2577
Publication Date:February 24, 2021
Abstract
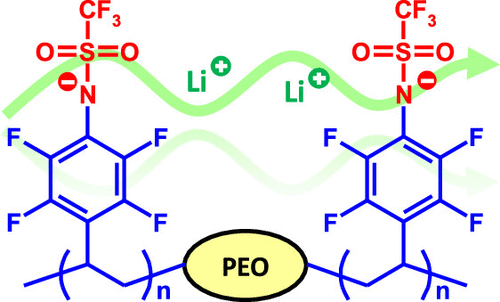
Solid single-ion conducting electrolytes based on well-defined block copolymers show great potential for use in lithium batteries. Here, we report on triblock copolymers with an ion-conductive poly(ethylene oxide) (PEO) center block and two flanking blocks of poly(lithium 2,3,5,6-tetrafluorostyrene-4-trifluoromethanesulfonamide). The copolymers were prepared through atom transfer radical polymerization (ATRP) of pentafluorostyrene using a bidirectional PEO macroinitiator, followed by quantitative nucleophilic aromatic substitution of p-fluorine atoms with sodium trifluoromethanesulfonamide. The ionic content of the copolymers was readily regulated by controlling the monomer feed ratio in the ATRP to obtain [EO]/[Li] between 4 and 88, and thermal decomposition occurred only above ∼300 °C, which indicates a high thermal stability. Above the melting point of the PEO center block, a copolymer containing 16 wt % of the flanking blocks ([EO]/[Li] = 40) reached a conductivity of 5.7·10–6 S cm–1 at 70 °C. The overall results indicate that well-designed polymers functionalized with lithium (N-perfluorophenyl)trifluoromethanesulfonamide groups show potential as solid single-ion conducting electrolytes.
1. Introduction
Solvent-free solid polymer electrolytes (SPEs) have during the past few decades been extensively studied as safe electrolyte materials for rechargeable lithium batteries. The main reason is their important advantages over standard liquid electrolytes, which include flame-resistance, improved life-time, high-temperature durability, increased mechanical flexibility, possibilities of low-cost design, and the function as a separator. (1−3) SPEs usually consist of a lithium salt (such as lithium bis[trifluoromethane]sulfonimide, LiTFSI) dissolved in a suitable polymer containing Lewis bases such as ether or carbonyl groups. Because the transport of the Li+ ions is strongly dependent on the segmental mobility of the coordinating polymer, the conductivity of SPEs is usually significantly below that of liquid electrolytes. In most studied SPEs, both the Li+ cations and the anions are mobile, which results in low cation transport numbers, typically between 0.1 and 0.5. (4−8) Another consequence of the dual ion mobility is the formation of concentration gradients during charge–discharge cycling of the cell, which in turn results in polarization effects such as voltage losses, high internal resistances, and adverse side reactions. (6−8) The salt concentration polarization effects are efficiently prevented by preparing single-ion conducting SPEs, where the anion is covalently linked to the polymer and only the “free” Li+ cations are able to move efficiently. (9−13) This means that the cation transference number approaches unity. (2) It is widely recognized that dramatic improvements in the battery performance can be achieved provided that single-ion conducting SPEs with sufficiently high conductivity can be designed and prepared. (12,14) In the last two decades, many different types of single Li+-conducting SPEs have been reported. (15−23) The most extensively studied are di- and triblock copolymers containing poly(ethylene oxide) (PEO) blocks combined with the lithium salt of poly(styrene trifluoromethanesulfonimide) PSTFSILi, first demonstrated by Bouchet et al. (24−28) Variations include the replacement of the PEO center block with polymethacrylates bearing short EO side chains (PPEOMA), (26,29) replacement of the PSTFSILi block with polymethacrylates bearing aliphatic trifluoromethanesulfonimide side chains (PTFSIMALi), (30) and use of a polymethacrylate backbone instead of a polystyrene one, for example, PPEOMA-b-PTFSIMALi. (31) In a comparative study by Devaux et al., it was found that PTFSIMALi-b-PEO-b-PTFSIMALi exhibited a higher conductivity than a corresponding triblock copolymer of PEO and PSTFSILi. (32) This was attributed to the lower glass transition temperature of PTFSIMALi compared to PSTFSILi.
In a previous work, we prepared triblock copolymers consisting of a PEO center block and two end blocks of the lithium salt of poly(2,3,5,6-tetrafluorostyrene-4-sulfonate) sPPFSLi. (33) Here, we report on the synthesis of single-ion conducting block copolymer SPEs with high Li+ conductivity functionalized with a lithium (N-perfluorophenyl)trifluoromethanesulfonamide [LiN(Pfp)(Tf)] salt. This salt has previously been prepared and electrochemically characterized by Huber and co-workers. (34) Both the electrochemical stability and ionic conductivity of LiN(Pfp)(Tf) solutions (0.1 M) in an ethylene carbonate:dimethyl carbonate (1:3 w/w) mixture were slightly lower than those of a corresponding LiTFSI solution but still sufficiently high for applications involving lithium-ion batteries. The high thermal stability and resistance against hydrolysis potentially makes LiN(Pfp)(Tf) an attractive substitute for LiPF6-based electrolytes at elevated temperatures. (34)
In the present work, BAB triblock copolymers with A-blocks of PEO and B-blocks of polypentafluorostyrene (PPFS) were first prepared by atom transfer radical polymerization (ATRP) of 2,3,4,5,6-pentafluorostyrene (PFS) from a PEO macroinitiator. In the next step, the PFS units were quantitatively transformed to (N-tetrafluorophenyl)trifluoromethanesulfonamide anions using a straightforward substitution reaction with the sodium trifluoromethanesulfonamide salt. The block ratio of the materials was conveniently controlled in the ATRP step to obtain the desired ionic content (expressed as the [EO]/[Li] ratio). The copolymers were evaluated as single Li+-ion conducting SPEs with a focus on the block copolymer structure, phase behavior, thermal stability, and conductivity. Future work will focus on further electrochemical characterizations and battery cell experiments.
2. Experimental Section
2.1. Materials
PEO (Sigma-Aldrich, Mn,SEC = 32.6 kg mol–1, Đ = 1.23), α,α′-dibromo-p-xylene (Sigma-Aldrich, 98%), copper(I) bromide (Sigma-Aldrich, 98%), 2,2′-bipyridyl (bipy, Acros, 99+%), anhydrous potassium carbonate (Sigma-Aldrich, 99 + %), 18-crown-6 (Sigma-Aldrich, 99+%), copper(0) powder (Sigma-Aldrich, 99.5%), sodium methoxide solution (Sigma-Aldrich, 25 wt % in methanol), hexane (Honeywell, 99%), diethyl ether (Honeywell, 99%), and N,N-dimethylacetamide (DMAc, Sigma-Aldrich, 99.8+%) were used as received. Tetrahydrofuran (THF, Honeywell, 99%) and o-xylene (Sigma-Aldrich, 99+%) were dried with molecular sieves (Acros, 4 Å 8–12 mesh). PFS (Sigma-Aldrich, 99%) was passed through a column of basic alumina (Alfa Aesar, activated, basic, Brockmann Grade I, 58 Å), degassed, and kept under an inert atmosphere. The ion-exchange resin Amberlite IR-120 in Na+ form (Sigma-Aldrich) was conditioned according to the instructions before use.
2.2. Synthesis of the PEO Macroinitiator
The Br-PEO-Br macroinitiator was prepared by functionalizing both chain ends of PEO with benzyl bromide-initiating groups. PEO (0.4 mmol, 14 g, 1 eq) was dissolved in a mixture of α,α′-dibromo-p-xylene (8 mmol, 2.1 g, 20 eq), 18-crown-6 (4 mmol, 1.5 g, 10 eq), dry potassium carbonate (8 mmol, 1.1 g, 20 eq), and THF (300 mL) in a dark brown round-bottom flask. The reaction mixture was refluxed under a nitrogen flow for 4 days at 80 °C. In the next step, the hot mixture was filtered to remove residual potassium carbonate. The filtrate was cooled to −18 °C and recrystallized twice in THF (300 mL). Finally, the product was collected as a white powder and dried under vacuum at 50 °C (yield: 97%).
1H NMR (400 MHz, CDCl3, ppm) δ 5.28 (s), 5.16 (s), 3.86–3.44 (broad).
2.3. Synthesis of Precursor Triblock Copolymers by ATRP
Four BAB triblock copolymers consisting of a central PEO A-block and flanking PPFS B-blocks were synthesized using Br-PEO-Br as a macroinitiator for ATRP of PFS (Scheme 1). In a representative polymerization, o-xylene (10 mL) was first degassed and heated to 80 °C followed by addition of Br-PEO-Br (0.1 mmol, 3.5 g, 1 eq), copper(0) powder (0.28 mmol, 18 mg, 2.8 eq), copper(I) bromide (0.4 mmol, 58 mg, 4 eq), and bipy (0.8 mmol, 126 mg, 8 eq). The mixture was degassed thrice at 40 °C under a nitrogen atmosphere before adding PFS via a syringe (Table 1). Next, polymerization was carried out at 110 °C for 48 h before it was terminated by adding THF (50 mL). The mixture was filtered through a column with alumina and then the copolymer was precipitated in hexane (200 mL). Finally, all the block copolymers were obtained as white powders after filtration. After a wash with diethyl ether, the samples were dried under vacuum at 50 °C for 50 h. These precursor block copolymers were denoted as PEO-PPFSx, where x is the PFS content in wt % as evaluated by 1H nuclear magnetic resonance (NMR) spectroscopy.

Table 1. Synthesis and Structure Data of the PEO-PPFSx Precursor Block Copolymers
| sample | [PFS]/[EO]a | monomer conversionb (%) | PPFS contentb (wt %) | degree of polymerization of PPFSb,c | PPFS block Mn,NMRb,c (kg mol–1) | Mn,NMRb,c (kg mol–1) | Mn,SECd (kg mol–1) | Đ MwMn-1d |
|---|---|---|---|---|---|---|---|---|
| PEO | 32.6 | 1.23 | ||||||
| Br-PEO-Br | 33.1 | 1.19 | ||||||
| PEO-PPFS5 | 8 | 73 | 5 | 9 | 1.8 | 36.8 | 34.7 | 1.29 |
| PEO-PPFS10 | 12 | 72 | 10 | 20 | 3.9 | 38.9 | 43.0 | 1.29 |
| PEO-PPFS26 | 30 | 75 | 26 | 63 | 12.3 | 47.3 | 45.1 | 1.39 |
| PEO-PPFS50 | 55 | 75 | 50 | 180 | 35.0 | 70.0 | 73.5 | 1.21 |
a
Molar ratio between the [PFS] monomer and [EO] units in the ATRP.
b
Calculated from the 1H NMR data.
c
Sum of both PPFS blocks, calculation based on Mn (PEO) = 35 kg mol–1.
d
Determined by SEC.
1H NMR (400 MHz, CDCl3, ppm) δ 3.9–3.4 (broad) and 3.0–1.9 (broad). 19F NMR (376 MHz, CDCl3, ppm) −143.1, −154.2, −161.1.
2.4. Preparation of BAB Triblock Copolymer Single-Ion Conductors
The PPFS blocks of the PEO-PPFSx triblock copolymers were functionalized with (tetrafluorophenyl)trifluoromethanesulfonamide anions to form TfnPPFS blocks. This was achieved by an aromatic nucleophilic substitution of the fluorine atoms at the para-position of the pentafluorophenyl rings using the trifluoromethanesulfonamide sodium salt (Scheme 1). The trifluoromethanesulfonamide sodium salt was first prepared by reacting trifluoromethanesulfonamide with a sodium methoxide solution (1:1 molar ratio) in anhydrous methanol followed by a wash with toluene. In a representative reaction, PEO-PPFS50 block copolymer (0.5 g, 1.29 mmol PFS, 1 eq) and trifluoromethanesulfonamide sodium salt (6.45 mmol, 1.1 g, 5 eq) were reacted in DMAc (20 mL) at 150 °C for 4 days to ensure complete substitution. Subsequently, the mixture was cooled to −18 °C before precipitating the product in hexane. The precipitate was collected by centrifugation, washed with cold DMAc and diethyl ether, and finally dried under vacuum at 50 °C for 24 h. Finally, the copolymers were transformed to the Li+ form by ion exchange and then dialyzed (MWCO = 3500 Da) in deionized water for 2 days to remove any trace impurities. The BAB triblock copolymer single-ion conductors were denoted as PEO-TfnPPFSLiy, where y denotes the TfnPPFS content in the copolymer expressed in wt %. The composition and molecular weights of the PEO-TfnPPFSLiy copolymers are listed in Table 1. The samples were first dried under vacuum at 60 °C overnight and then further dried in the melt state while stirring under high vacuum (0.2 Pa) at 80 °C for 48 h, before being stored in an inert atmosphere.
19F NMR (376 MHz, DMSO-d6, ppm) −77.5, −139.3, −142.8.
2.5. Characterization
The structures of all polymer samples were analyzed and confirmed by 1H NMR spectroscopy, employing a Bruker DRX400 spectrometer and DMSO-d6 or CDCl3 as a solvent. The block copolymers were further characterized by 19F NMR spectroscopy.
The number average molecular weight (Mn) and polydispersity (Đ) of the PEO precursors and the precursor block copolymers were measured by size-exclusion chromatography (SEC) using a Viscotek GPCmax VE-2001 instrument. Samples dissolved in chloroform were analyzed using three Shodex columns (KF-805, KF-804, and KF-802.5) and a refractive index detector at an elution rate of 1 mL min–1. Four different PEO standards from Agilent with Mn = 4.25, 12.6, 50, and 100 kg mol–1 were used for the calibration. The Mn value of the commercially available PEO precursor was determined to be 32.6 kg mol–1. In addition to the Mn values of the precursor triblock copolymers measured directly by SEC (MnSEC), presumably more accurate Mn values were calculated based on the Mn of the PEO block determined by SEC and their PFS content determined by 1H NMR analysis (MnNMR,Table 1).
A TA Instruments Q500 TGA analyzer was employed for thermogravimetric analysis (TGA) of the polymers. The thermal degradation was studied at a heating rate of 10 °C min–1 up to 600 °C under a nitrogen atmosphere. Before this analysis, the samples were dried at 130 °C for 10 min. The thermal degradation temperature (Td,95) was taken as the point where 95% of the sample mass remained. Differential scanning calorimetry (DSC) was carried out using a TA Instruments Q2000 calorimeter at a heating/cooling rate of 10 °C min–1 under a nitrogen atmosphere. The precursor triblock copolymers as well as samples PEO-TfnPPFSLi16 and PEO-TfnPPFSLi37 were analyzed during the temperature cycle: 150 → −60 → 150 °C. The samples in the PEO-TfnPPFSLiy series with the lowest and highest PEO contents (i.e., PEO-TfnPPFSLi8 and PEO-TfnPPFSLi63) were analyzed from −80 °C (the lowest possible temperature with the DSC setup) in an attempt to detect glass transitions at lower temperatures. The melt (Tm) and crystallization (Tc) temperatures were determined during the heating and cooling scans, respectively.
The ionic conductivity of the BAB triblock copolymer single-ion conductors was determined by measuring the impedance spectra during a heating–cooling–heating cycle between 0 and 90 °C. Inside an Ar-filled glovebox, dried electrolyte samples with a diameter of 15 mm and a thickness of approximately 105 μm were sandwiched between two gold-plated brass coin electrodes separated by a PTFE ring. A computer-controlled Novocontrol BDC40 high-resolution dielectric analyzer coupled to a Novocool cryostat unit was used in the analysis. Samples were measured between 10–1 and 107 Hz at a 50 mV AC amplitude. Subsequently, the DC conductivity values were determined using the Novocontrol software WinDETA.
3. Results and Discussion
3.1. Molecular Design and Synthesis
Four PEO-TfnPPFSLiy single-ion conducting BAB triblock copolymers with different values of y, that is, with different block lengths and [EO]/[Li] ratios were prepared in accordance with Scheme 1. The materials were based on a precursor PEO block with Mn = 33 kg mol–1 and reasonably low polydispersity (Đ) with MwMn–1 = 1.23 (Table 1). The difunctional Br-PEO-Br macroinitiator for the ATRP of the PPFS blocks was prepared by chain-end functionalization through a reaction of the terminal hydroxyl groups with α,α′-dibromo-p-xylene mediated by potassium carbonate in THF at 80 °C (Scheme 1). In order to ensure full functionalization and to avoid chain extension reactions, dibromoxylene was used in a 10-fold excess. In addition, the solvation of potassium carbonate in THF was facilitated by the presence of 18-crown-6. Figure 1a shows the 1H NMR spectrum of the product, which contained shifts of the benzyl bromide protons at 5.28 ppm and of the benzyl ether protons at 5.16 ppm with equal integrals (within the error of the method). This confirmed the successful chain-end modification and indicated the absence of any significant degree of chain extension during the reaction. Furthermore, the SEC analysis of PEO and Br-PEO-Br showed that the values of Mn and Đ remained virtually unaffected after the chain-end functionalization, which again implied efficient suppression of chain extension reactions (Table 1, chromatograms included in Figure S1).

In the synthesis of the PEO-PPFSx precursor block copolymers by ATRP, the degree of polymerization of the individual PPFS blocks was targeted to between 9 and 180 by controlling the molar ratio of PFS monomer to Br-PEO-Br macroinitiator. This was done in order to obtain final PEO-TfnPPFSLiy copolymers with [EO]/[Li] ratios in the range between approx. 4 and 88 (Tables 1 and 2). The ATRP of PFS was carried out using Br-PEO-Br at 110 °C by employing a CuBr/bipy system in o-xylene (Scheme 1). (33) During the polymerization, Cu(I) was regenerated in the presence of a catalytic amount of Cu(0) powder. The commonly employed molar ratio of PhCH2Br:Cu(I)Br:bipy equal to 1:1:2 produced unrepeatable polymerization results, a possible result of the high molecular weight macroinitiators. Instead, we found that a ratio of 1:2:4 gave reproducible results and products.
Table 2. Synthesis and Structure Data of the PEO-TfnPPFSLiy Block Copolymers
| sample | block copolymer precursor | TfnPPFSLi content (wt %) | TfnPPFSLi block Mna (kg mol–1) | Mn (kg mol–1) | [EO]:[Li]b |
|---|---|---|---|---|---|
| PEO-TfnPPFSLi8 | PEO-PPFS5 | 8 | 3.0 | 38.0 | 88 |
| PEO-TfnPPFSLi16 | PEO-PPFS10 | 16 | 6.6 | 41.6 | 40 |
| PEO-TfnPPFSLi37 | PEO-PPFS26 | 37 | 20.7 | 55.7 | 12 |
| PEO-TfnPPFSLi63 | PEO-PPFS50 | 63 | 59.2 | 94.2 | 4 |
a
Sum of both TFnPPFSLi blocks.
b
Molar ratio of [EO] to [Li].
The 1H NMR analysis of the purified products confirmed the formation of the block copolymers, and the content of the PEO-PPFSx precursor block copolymers was calculated by taking into account the signal intensities of the PPFS and PEO blocks found at 1.9–3.0 ppm and 3.4–3.9, respectively (Figure 1b). (33) Next, the degree of polymerization of the PPFS blocks was calculated by considering the PPFS content and Mn of the PEO precursor. The monomer conversion was found to be between 72 and 75% by comparing the obtained degree of polymerization with the theoretical one reached at full conversion (Table 1). In addition, the 19F NMR spectrum showed the presence of expected signals from the fluorine atoms in the PPFS blocks (o-F at −143, m-F at −161, and p-F at −154 ppm), which again demonstrated the successful ATRP reaction (Figure 1c). (33) A comparison of the Mn values of the block copolymers measured using the SEC analysis (chromatograms included in Figure S1) and the values calculated from Mn of the PEO precursor and the NMR data of the respective PEO-PPFSx precursor block copolymers showed little difference (Table 1). Also, the SEC measurements of the block copolymers revealed polydispersities around Đ = 1.3, which is in the range expected for polymers produced by ATRP.
In order to obtain PEO-TfnPPFSLiy copolymers, the PPFS blocks of PEO-PPFS were modified via substitution of the para-fluorine atoms by a reaction with the sodium trifluoromethanesulfonamide salt to form the (N-tetrafluorophenyl)trifluoromethanesulfonamide anions (Scheme 1). This salt was prepared separately by deprotonating trifluoromethanesulfonamide using sodium methoxide. An excess of the trifluoromethanesulfonamide sodium salt was added to DMAc solutions of the block copolymers in the substitution reaction. To obtain the final product, the sodium counterions of the reaction products were exchanged with lithium ions using a standard ion-exchange procedure. In order to remove all traces of any free salt, the PEO-TfnPPFSLiy samples were dialyzed in deionized water for 2 days. Subsequently, all samples were dried under high vacuum at 80 °C. Figure 1d shows a representative 19F NMR spectrum of the PEO-TfnPPFSLiy series. As can be seen, the spectrum showed a strong signal at −77.5 ppm originating from the −CF3 groups and two very weak signals arising from the ortho-F and meta-F at −142.8 ppm and −139.3 ppm, respectively. In addition, the signals from the pentafluorophenyl ring were absent. Within the accuracy of the method, this confirmed the complete and selective introduction of the (N-tetrafluorophenyl)trifluoromethanesulfonamide anions (Figure 1). (33) Hence, the calculation of the ionic content of these materials was based on the complete substitution of the para-fluorine atoms of the PPFS blocks.
3.2. Thermal Stability
The thermal decomposition of the PEO-TfnPPFSLiy copolymers and the various precursor polymers was investigated by TGA under a nitrogen atmosphere. PPFS and PEO homopolymers decomposed in single steps at Td,95 = 418–430 (35,36) and 363 °C, respectively. The TGA profile of nonionic PEO-PPFS5 showed a single decomposition step at Td,95 = 342 °C. When the PPFS content was increased to x = 10 wt % and above, the value of Td,95 increased to 355–358 °C and a second decomposition step appeared at ∼420 °C and increased with x (Table 3, Figure S2). The two steps were likely the result of the decomposition of the PEO and PFS blocks, respectively. After the introduction of the (N-tetrafluorophenyl)trifluoromethanesulfonamide anions in the PEO-TfnPPFSLiy series, the value of Td,95 decreased in relation to the corresponding precursor copolymer in the PEO-PPFSx series (Table 3, Figure 2). Moreover, the decomposition of the former copolymers occurred in one step. The Td,95 value of the ionic block copolymers was close to Td,95 = 350 °C at low and moderate ionic contents (i.e., y = 8–26 wt %) but decreased to 312 °C for sample PEO-TfnPPFSLi63. Hence, it was likely that the decomposition of PEO-TfnPPFSLiy started through the decomposition of the ionic groups. The block copolymers exhibited sufficient thermal stability for most electrolyte applications (i.e., Td,95 ≫150 °C). (4)

Table 3. Thermal Data of the Precursor Polymers and the PEO-TfnPPFSLiy Block Copolymers
| sample | Td,95 (°C) | Tc (°C) | ΔHca (J g–1) | Tm (°C) | ΔHma (J g–1) |
|---|---|---|---|---|---|
| PEO | 363 | 44 | 154 | 65 | 157 |
| PEO-PPFS5 | 342 | 46 | 130 | 61 | 152 |
| PEO-PPFS10 | 358 | 43 | 158 | 65 | 162 |
| PEO-PPFS26 | 357 | 49 | 187 | 63 | 191 |
| PEO-PPFS50 | 355 | 50 | 164 | 64 | 190 |
| PEO-TfnPPFSLi8 | 349 | 41 | 170 | 63 | 174 |
| PEO-TfnPPFSLi16 | 352 | 46 | 177 | 66 | 181 |
| PEO-TfnPPFSLi37 | 353 | 40 | 198 | 63 | 204 |
| PEO-TfnPPFSLi63 | 312 | 35 | 208 | 60 | 214 |
a
Calculated based on the PEO weight.
3.3. Phase Transitions
The phase behavior of the different polymers and copolymers was studied by DSC and the data are listed in Table 3. Crystallization and melting of the PEO precursor block occurred at Tc = 44 °C and Tm = 65 °C, respectively. PPFS has a Tg slightly above 100 °C (101–105). (35,37) However, analysis of the PEO-PPFSx series revealed only PEO crystallization and melt transitions during cooling and heating, respectively, and no glass transition above −80 °C. Tc was found in the range of 43–50 °C, that is, generally slightly higher than for PEO, while Tm was very close to that of PEO, 61–65 °C. The slightly higher Tc compared to neat PEO could be due to an orientation effect on the PEO blocks by the PPFSx domains. Based on the PEO content, the heat of crystallization (ΔHc) was lower for PEO-PPFS5 than for neat PEO but then increased at higher PPFS contents (Table 3, Figure S2) until PEO-PPFS50, which showed a decrease in ΔHc compared with PEO-PPFS26. The heat of melting (ΔHm) values in the series increased with x until reaching approximately 120% of the neat PEO for samples PEO-PPFS26 and PEO-PPFS50. Again, the higher crystallinity of the PEO blocks may be the result of an orientation induced through the connectivity with the PPFS domains in the microphase-separated block copolymer. The propensity for microphase separation likely increased with the length of the PPFS blocks, which led to a higher degree of segment orientation, crystallization temperature, and crystallinity of the PEO blocks as the weight fraction of PPFS increased. In all cases, ΔHm > ΔHc, which indicated the occurrence of some cold crystallization, and this was most pronounced for sample PEO-PPFS50.
The DSC traces of the PEO-TfnPPFSLiy series also did not show any glass transitions (Figure 3) above −80 °C. Both Tc and Tm were found close to those of the neat PEO for all samples. In contrast, the values of ΔHc and ΔHm increased consistently with x until PEO-TfnPPFSLi63 reached approximately 136% of the respective value of the neat PEO, in a similar way as observed for the PEO-PPFSx series (Table 3). Normally, the crystallinity of PEO decreases with the salt content due to the interactions between Li+ and EO. (38,39) The observed discrepancy might be due to the low compatibility between TfnPPFS– and PEO due to the rigid structure of the former. (40) This may lead to the formation of two distinct phases with one Li+-rich TfnPPFS– and one PEO phase with a low Li+ concentration, where the crystallization was facilitated by the orientation of the PEO blocks.

3.4. Ionic Conductivity
The ionic conductivity of the block copolymers in the PEO-TfnPPFSLiy series was measured by electrochemical impedance spectroscopy. In these single-ion conducting triblock copolymers, the Tfn anions were covalently attached to the TfnPPFS blocks, which were consequently restricted to exclusively short-range segmental mobility. Presumably, only the Li+ cation had long-range mobility in the materials and the transport number was expected to be very close to unity. Furthermore, we anticipated that the dissociation of the lithium ions was greatly facilitated by their position in between the strongly electron-withdrawing tetrafluorobenzene and Tf units. The Li+ conductivity of PEO electrolytes is normally coupled to the segmental mobility. Hence, any crystallinity is likely to decrease the level of conductivity.
The DC conductivity values of the samples were determined from the frequency-independent conductivity plateaus observed in the plots of AC conductivity versus frequency (Figure S3). The conductivity data measured during heating of the PEO-TfnPPFSLiy samples from 0 to 90 °C are shown in Figure 4. As seen, the conductivity of all samples increased sharply from low levels below 70 °C because of melting of the PEO blocks. Consequently, the conductivity of these samples increased by almost five orders of magnitude between 0 and 90 °C. At temperatures above the melting point of the PEO center blocks (i.e., above 60 °C), PEO-TfnPPFSLi16 with [EO]/[Li] = 40 clearly exhibited the highest conductivity (5.7·10–6 and 9.7·10–6 S cm–1 at 70 and 90 °C, respectively) in the sample series (Figure 5). At higher ionic contents (i.e., lower values of [EO]/[Li]), the ionic conductivity decreased, which was likely due to the decreased mobility of the PEO segments. On the other hand, PEO-TfnPPFSLi8 with [EO]/[Li] = 88 exhibited a lower ionic conductivity due to the lower charge carrier concentration.


The ionic conductivities may be compared to values obtained from the triblock copolymers of pentafluorostyrene lithium sulfonate (sPPFSLi) and PEO. (33) Here, a PEO-b-sPPFSLi polymer with [EO]/[Li] ≈ 18 exhibited the highest ionic conductivity at temperatures above the melting point of the PEO block. This polymer achieved ionic conductivities of 2.0·10–5 and 3.0·10–5 S cm–1 at 70 and 90 °C, respectively. Hence, more than three times higher values for PEO-TfnPPFSLi16 were obtained. Two further polymers from a former study with [EO]/[Li] = 40 and 10, respectively, reached ionic conductivities of 2·10–6 and 3·10–6 S cm–1 at 90 °C. This indicates that a significantly higher conductivity may be reached by PEO-TFnPPFSLiy samples prepared with [EO]/[Li] in the range between 12 and 40. Bouchet et al. have previously prepared triblock copolymers having a center block of PEO (35 kDa) flanked by two PSTFSILi blocks. (24) They reported a conductivity of approximately 2·10–5 S cm–1 at 70 °C for the polymer with [EO]/[Li] = 36, which was also achieved in a later study. (32) This is similar to the conductivity reached by a PTFSIMALi-b-PPEOMA diblock copolymer with [EO]/[Li] = 6 prepared by Porcarelli et al. (31)
4. Conclusions
A series of triblock copolymers with a PEO center block flanked by two TfnPPFSLi blocks was readily prepared by ATRP of PFS using a PEO macroinitiator followed by quantitative substitution of p-fluorine using trifluoromethanesulfonamide. The ionic content was conveniently varied by controlling the amount of PFS in the monomer feed in the ATRP. Thermal decomposition occurred only above 300 °C, which indicated high thermal stability of the copolymers. The crystallization of the PEO center block was not significantly disturbed by the two flanking blocks, indicating incompatibility between the blocks. At temperatures below the melting point of the PEO center block (i.e., below ∼60 °C), the ionic conductivity remained low. However, at 70 °C, the ionic conductivity reached rather high values, approaching 6·10–6 S cm–1. The present results indicate that polymers functionalized with lithium (N-perfluorophenyl)trifluoromethanesulfonamide groups show promising potential as solid single-ion conducting electrolytes. Future work will focus on studies of electrochemical stability and enhancing the conductivity by optimizing the [EO]/[Li] ratio and employing alternative noncrystallizing ion-conductive blocks.
【Article link】
https://doi.org/10.1021/acsaem.0c03141